
It looks like your browser is incompatible with our website.
If you are currently using IE 11 or earlier, we recommend you update to the new Microsoft Edge or visit our site on another supported browser.
If you are currently using IE 11 or earlier, we recommend you update to the new Microsoft Edge or visit our site on another supported browser.


The information you need to make meaningful discoveries is hard to find – or at least it was.
ResoluteAI's secure research platform lets you search aggregated scientific, regulatory, and business databases simultaneously. Combined with our interactive analytics and downloadable visualizations, you can make connections that lead to breakthrough discoveries.
Competitive intelligence |
Drug repurposing |
Institutional knowledge retrieval |
Key Opinion Leader identification |
Market research |
Pharmacovigilance |
Post market surveillance |
Regulatory risk reduction |
Repurposing of past research |
Technology landscaping |
Technology transfer |
White space analysis |


Nebula, ResoluteAI's enterprise search product for science, makes your proprietary information easily findable and accessible.
Learn more about Nebula.
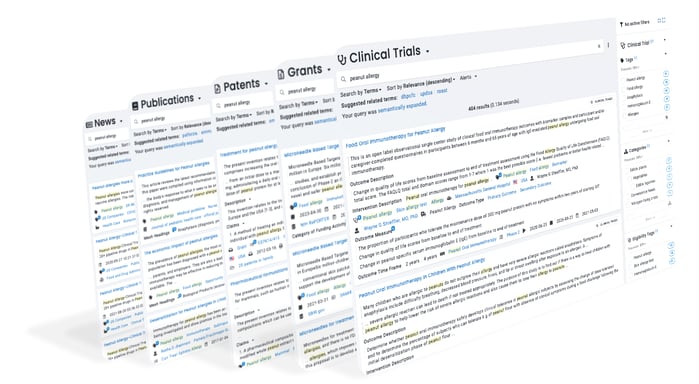
Foundation is a secure research platform that provides exploration, search, analytics, and alerts on over a dozen science-focused databases.
Learn more about Foundation.
.svg) Natural Language Processing
Natural Language ProcessingStructured metadata created from unstructured text, semantic expansion, conceptual search, and document similarity search.

Filter results based on structured metadata created by NLP or contained in the source database.

ResoluteAI’s proprietary science-focused ontology plus PubChem, MeSH, and more.

Save searches and have results delivered directly to your inbox.

Export your search results for analysis outside the ResoluteAI platform.

Index and understand text that appears in handwritten documents, images, and video.

Open documents embedded in other documents for complete indexing and discovery.

Identifies images, graphs, charts, and logos in documents and video.

Transcribes and indexes talk tracks from webinars, videos, and other presentations.

Ingestion and indexing works on dozens of formats including Microsoft Office documents, PDFs, .msg, audio, and video.

SharePoint, Azure Blob, Box, DropBox, GDrive, and many other data storage solutions are supported.
.png?width=840&height=411&name=MicrosoftTeams-image%20(75).png)
"Nebula connected me in a very strategic way… What took me a week just to put in subfolders would come out in less than a couple seconds on the platform. It was phenomenal, incredible." - Dr. Dolly Tyan, Aditx Therapeutics Inc.

If you spend more than a couple of hours a week looking for academic research, or scanning through patents, or exploring tech transfer opportunities, a research platform that brings all these databases together, and more, might be helpful. Maybe you have research organization tools, or use desktop software to organize research for yourself or for your colleagues. Online research platforms that bring all your results together might save you a tremendous amount of time. And money. Information platforms exist for all domains, ResoluteAI’s online research platform is focused on scientific research and is used by professionals in pharma, biotech, chemical, diagnostics, consumer products, and many other industries around the world.
See how ResoluteAI's research platform can help you. Book a demo today.
